Electron microscopy (EM) data is getting pretty big in the bioinformatics world at the moment. Whilst I'm not familiar with the machinery and physics behind it (I'm sure I'll take a look at some point), you are left with a field or volume with certain densities in certain places. This gives a rough outline of the object you are trying to map out - in our case, proteins.
Imagine you have a rough model of your protein, possibly modelled using another method like X-ray crystallography. If you have a shell or shape to fit in inside, you can sort of squish it into place and get a more accurate model. It's a second set of data that helps you be more sure you've got the right model in the first place.
At the moment, the accuracy of x-ray crystallography exceeds that of EM (around 2 angstrom resolution compared with 6 to 10 Angstroms, more or less). However, it's one of these technologies that is getting better all the time.
UCSF's Chimera is one of the major programs for working with proteins, fields and all manner of structural biology. It's an impressive piece of software to be sure, though it's interface is showing it's age. That's why the team over in San Francisco is working on ChimeraX. This is a new version which looks much slicker. It's still in beta though, with many of the features not implemented yet.
ChimeraX has a more streamlined interface for dealing with plugins. The ChimeraX Toolshed is an online repository of several plugins that users can download. If you have a Google account and gain permission, you can submit plugins here too.
ChimeraX uses Python 3 throughout. It has a few bindings to several C libraries here and there (I believe the rendering is performed in OpenGL at the moment). There exists a reasonable API and a good development guide document for many of the functions within ChimeraX, though the objects and the data they contain are not expanded on. I suspect this is more of a Python thing if I'm honest.
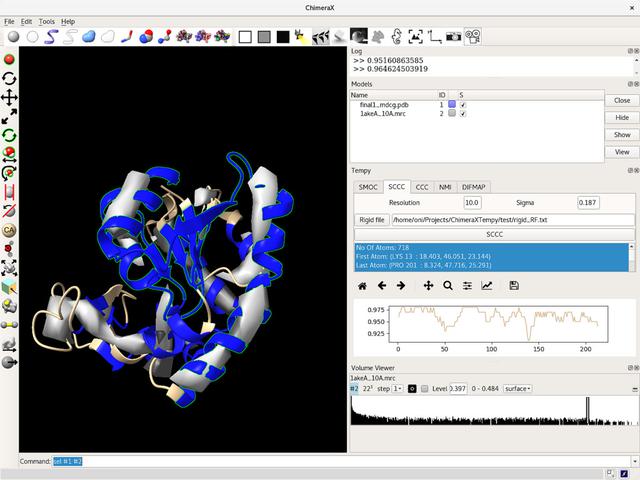
Tempy is a set of Python scripts written by Agnel Joseph and Maya Topf (and others) at Birkbeck University of London. They provide fitting and scoring functions for EM data. The scripts are written in Python 2.6, and rely on the BioPython libraries. At present, documentation for Tempy is limited and the API docs for BioPython are light in some areas.
My task was to make Tempy work with ChimeraX - making it easy for users to work with their own EM datasets.
ChimeraX has a good set of documents but when you really get into it, you quickly find yourself a bit lost. To begin with, Tempy requires it's models in the BioPython format, and it's Maps in the MRC format. ChimeraX has it's own ideas about models and volumes. While it's reasonably easy to track these classes down, what data they contain is more tricky. You find yourself writing the following a lot:
print(dir(session))
... or something similar. Figuring out exactly what a ChimeraX model contains can be a lengthy process. It's possible, once you've figured out which class is doing what, to take a look at the source-code and go from there. Eventually, I managed to get a rough conversion working from ChimeraX to BioPython, but it's still not perfect. Likely, without speaking to the developers a little more, understanding the different terminology and some more of the biology, it's unlikely I'll get a complete 100% conversion, but this is close enough for the scoring to work.
atomicMasses = {'H':1, 'C': 12, 'N':14, 'O':16, 'S':32}
def chimera_to_tempy_atom(atom, serial):
""" Convert one of the ChimeraX atoms to a Tempy BioPython Style one """
ta = BioPyAtom([])
ta.serial = serial
ta.atom_name = atom.name
ta.alt_loc = atom.alt_loc
ta.res = atom.residue.name
ta.chain = atom.chain_id
ta.res_no = atom.residue.number
ta.model = atom.chain_id # PDB number?
ta.init_x = atom.coord[0]
ta.init_y = atom.coord[1]
ta.init_z = atom.coord[2]
ta.bfactor = atom.bfactor
ta.x = atom.coord[0]
ta.y = atom.coord[1]
ta.z = atom.coord[2]
ta.occ = atom.occupancy
ta.temp_fac = atom.bfactor
ta.elem = atom.element.name
ta.charge=""
ta.record_name = "HETATM"
if atom.in_chain:
ta.record_name = "ATOM"
#Mass of atom as given by atomicMasses global constant. Defaults to 1.
ta.mass = atom.element.mass
ta.mass = atomicMasses.get(ta.atom_name[0])
ta.vdw = 1.7
ta.isTerm = False
ta.grid_indices = []
return ta
Many of the functions look like this - simply copying from one data-structure to another. There was a lot of trial and error involved!
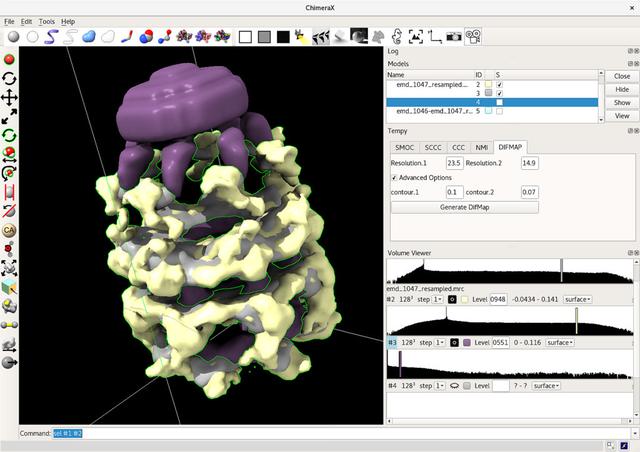
The Tempy developers are sticking with Python 2 at the moment. With Chimera only supporting Python 3, I've had to make judicious use of the program:
2to3
But this isn't quite enough. In Python 2, you could often assume that certain functions would return a List or an Iterable. In Python 3 that is not the case. Quite often you'll get a Map object or similar and you'll need to do something like:
#origin = map(float, first_line[1:4])
origin = list(map(float, first_line[1:4]))
2to3 won't change these sorts of things (I mean, how could it really?) so one needs to make these changes manually, every-time the Tempy code base changes. At some point I'll write some diff files and automatically apply them after a run through with 2to3.
Qt is perhaps the GUI-Library-du-Jour for a large number of projects. I suppose there are others, but nothing seems as complete and cross platform as Qt is. ChimeraX uses PyQt - a bindng to the QT libraries for Python.
User-interface programming is perhaps the worst kind of programming, just after writing tests! The reason? It's mostly copying and pasting long winded statements and linking signals to receivers, actions to callbacks or whatever the library designer decided to call such things that week. You end up with lines upon lines of very similar code that requires no thought in terms of problem solving or clever programming. I recommend some good YouTube videos to watch while you write this sort of thing.
Tests are perhaps the most boring but one of the most important parts of software design. I admit, it feels like eating nothing but the crusts of brown bread because it's good for you. Nevertheless, testing things has been very useful. There are quite a few different testing strategies and frameworks. The one I settled for in Python was unittest which has the usual setup of create a class that extends another, override a couple of functions and voila! You have a test! Something a little like this:
import unittest
class MyTest (unittest.TestCase):
def setUp(self):
# setup the data what not
pass
def tearDown(self):
# reset the state
pass
def test_myTest(self):
# Perform the actual test
Because I'm converting code from one version and place to another writing tests is both easier and more vital; the numbers that the new code produces, given the input numbers should match the ones that the original version produces.
So far, the plugin is still in the beta phase. I'm currently moving over functions that compare maps and create new ones inside ChimeraX. Hopefully, there will be some happy users and new screenshots I can post here.
