Some of you may know that I've been studying bioinformatics at Birkbeck, University of London. I've got lots of reasons why that I might discuss in another post, but I'd like to talk about some of the basics of Machine learning, structural biology and some tips about how one can go about studying for an MRes. The research path is long and difficult but very rewarding. If you are doing your job right, you'll discover something no-one else has ever discovered, even if that thing is really tiny. There are few callings in life that can compete with that.
Biology is a big subject - and I mean really big! In terms of raw data, I heard a factoid that the data output from somewhere like EMBL is much larger than CERN. The problem is not just about volume though. Biological data is extremely heterogeneous. Sometimes it's empirical and nicely formatted. Other-times it's qualitative and messy. There are so many formats and workflows to keep track of, I'm amazed that anything gets done at all. Indeed, this is a problem that biology has when it comes to how certain we can be of any results. A recent talk at the CCC highlights the problem with P-Values. Biology has typically relied on a 0.05 P value which is frankly, terrible. Remember that whole 5 Sigma thing that was all the rage when the Higgs Boson was found? Physicists there could afford to be accurate. In biology, the systems are so much more complicated; drawing even a correlation, let alone a causation appears to me to be much harder.
As computers and the systems we build become more and more complex, I've noticed a striking resemblance between the two. When we take artificial intelligence into account, we are building deeply complicated, power hungry systems that are beginning to defy reductionism and end-to-end, logical reasoning. The two fields are, in my mind, becoming a lot closer than I had previously thought.
Structural biology is where I've been focusing my attention. Lots of people think that DNA is where it's at in biology. I respectfully disagree. DNA is all about statistics, matching endless streams of letters and working around crappy software and slow machinery. Wouldn't you rather take apart and build tiny machines made of amino acids? Of course you would! Structural biology has many similarities with computer graphics funnily enough. At one point in time, the fastest renderer of spheres was written by a biologist! There are many algorithms related to matching structures, converting between co-ordinates and all sorts of linear algebra that I feel quite at home with.
Benjamin's top tip! If you love computer graphics, programming and want to make a difference, go into biology!

One thing biologists love to do, is predict what might happen given a certain starting condition. You've probably heard of Folding at Home right? Protein folding is a really important and really difficult problem. In a nutshell, you have a known list of amino acids like Tyrosine, Glycine, Proline, Glycine etc and you want to know what structure you'll end up with at the end. This 3D structure defines what the protein will actually do. If we know what a particular sequence will turn into, we can figure out what a certain DNA sequence will result in. We can create better drugs or delete problematic DNA errors. It's a big thing.
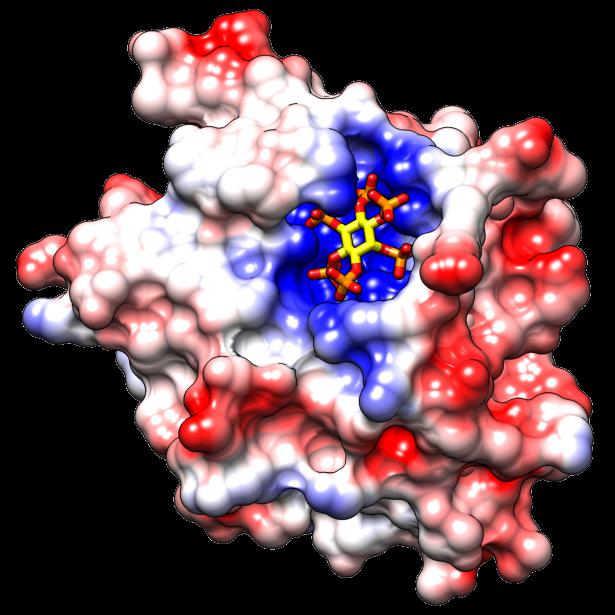
My area looks at a similar, but smaller area. I'm thinking if it's possible to model a certain class of loops - the one's on antibodies specifically. Given a sequence of Amino Acids, can we predict what shape the loop will have? If we can, we can make better antibodies. Antibodies are great because they can be used as markers and drugs.
Deep learning has been the hot, sexy topic for quite a while now. It's funny how an old idea can suddenly come back and take the world by storm. Biologists have been using neural nets for some time now. PSIPRED is a is a good example of a program I've used before that uses neural nets. It tries to detect secondary structure when given an amino acid sequence. The neural networks in the news are typically image and classifier related and the things they can do are quite amazing. The canonical example is the Googlenet based on the work of Lecun et al.
Such nets are called deep, not only because they have several layers, but also because they rely on convolutions. A convolution operation takes a kernel of a certain size and convolves it over the input. The kernel might be, say, an 8 x 8 patch that sums up all the values within it. reducing the input by a factor of 8 (roughly). However, at the same time, it creates a third dimension that only gets deeper in time. For instance, an input image might be 256 x 256 in size, made up of single values. After the first convolution it might be 128 x 128 x 3 depending on your kernel and it's operation. A further operation might reduce further to 64 x 64 x 6. The last dimension keeps getting deeper as the original dimensions get smaller.
Google released Tensorflow sometime ago, and I used it briefly when I was looking at Natural Language Processing. This time around I thought I'd learn a little more first. I'd recommend the coursera introduction to machine learning. You'll get the bare basics which you'll need to get started. I decided to use Tensorflow over Caffe or Keras because it's easy to setup and there's plenty of documentation. Not only that, it's pitched at just the right level - it's quite powerful but quick to use. I decided to setup a machine to run all of my nets on. I managed to get hold of a machine being thrown out from the University. With a spare graphics card kicking around I had a machine I could install Arch Linux on and take advantage of Tensorflow's GPU support. Tensorflow has support for a few languages, the most popular being Python (which is what I use). It's good to know that C is also an option for a more final product.
Tensorflow has the idea of the graph and the session. Tensors are supposed to flow through the graph in a session. You build a graph that is made up of operations that pass tensors around. With this graph built, you create a session which takes some inputs and runs them through the graph. Hack-a-day has a really good introduction to Tensorflow that describes how the classic idea of a set of neurons and links, maps on to a series of matrix multiplications. Essentially, most tensors we are likely to deal with are either matrices (2D) or maybe 3D.
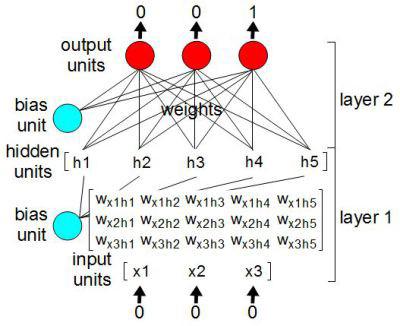
The following is a short example of one of my early test nets. This example creates a convolutional neural net that has 1 convolutional layer and two fully-connected layers. At points, I resize the tensors to perform certain operations, but mostly, it's a series of matrix multiplications and additions. Finally, I use a tanh activation function. If you read a lot of the Tensorflow examples out there, you'll see a lot of ReLUs being used, but for our purposes, we need a nice range between -1 and 1.
graph = tf.Graph()
with tf.device('/gpu:0'):
with graph.as_default():
tf_train_dataset = tf.placeholder(tf.bool,
[None, FLAGS.max_cdr_length, FLAGS.num_acids],name="train_input")
output_size = FLAGS.max_cdr_length * 4
dmask = tf.placeholder(tf.float32, [None, output_size], name="dmask")
x = tf.cast(tf_train_dataset, dtype=tf.float32)
W_conv0 = weight_variable([FLAGS.window_size,
FLAGS.num_acids, FLAGS.num_acids] , "weight_conv_0")
b_conv0 = bias_variable([FLAGS.num_acids], "bias_conv_0")
h_conv0 = tf.tanh(conv1d(x, W_conv0) + b_conv0)
dim_size = FLAGS.num_acids * FLAGS.max_cdr_length
W_f = weight_variable([dim_size, output_size], "weight_hidden")
b_f = bias_variable([output_size], "bias_hidden")
h_conv0_flat = tf.reshape(h_conv0, [-1, dim_size])
h_f = tf.tanh( (tf.matmul(h_conv0_flat, W_f) + b_f)) * dmask
W_o = weight_variable([output_size, output_size], "weight_output")
b_o = bias_variable([output_size],"bias_output")
y_conv = tf.tanh( ( tf.matmul(h_f, W_o) + b_o) * dmask, name="output")
return graph
With this graph in place, I can then run it over on my GPU with the following session:
def run_session(graph, datasets):
''' Run the session once we have a graph, training methodology and a dataset '''
with tf.device('/gpu:0'):
with tf.Session(graph=graph) as sess:
training_input, training_output, validate_input, validate_output, test_input, test_output = datasets
# Pull out the bits of the graph we need
ginput = graph.get_tensor_by_name("train_input:0")
gtest = graph.get_tensor_by_name("train_test:0")
goutput = graph.get_tensor_by_name("output:0")
gmask = graph.get_tensor_by_name("dmask:0")
stepnum = 0
# Working out the accuracy
basic_error = cost(goutput, gtest)
# Setup all the logging for tensorboard
variable_summaries(basic_error, "Error")
merged = tf.summary.merge_all()
train_writer = tf.summary.FileWriter('./summaries/train',graph)
train_step = tf.train.GradientDescentOptimizer(FLAGS.learning_rate).minimize(basic_error)
tf.global_variables_initializer().run()
while stepnum < len(training_input):
item_is, item_os = next_item(training_input, training_output, FLAGS)
mask = create_mask(item_is)
summary, _ = sess.run([merged, train_step],
feed_dict={ginput: item_is, gtest: item_os, gmask: mask})
if stepnum % 100 == 0:
mask = create_mask(validate_input)
train_accuracy = basic_error.eval(
feed_dict={ginput: validate_input, gtest: validate_output, gmask : mask})
print('step %d, training accuracy %g' % (stepnum, train_accuracy))
train_writer.add_summary(summary, stepnum)
stepnum += 1
# save our trained net
saver = tf.train.Saver()
saver.save(sess, 'saved/nn02')
There are a few little gotchas here that are worth mentioning. It's import to call:
tf.global_variables_initializer().run()ginput = graph.get_tensor_by_name("train_input:0")There's more to this example that I've not included, such as the cost functions and various support functions to create and initialise proper weights but I think we can agree that it's not a lot of code to generate a fully usable neural network.
These of you who have done a research based masters degree or a PhD will no doubt have your own war stories. I'm sure there has been plenty written about the research experience but although I'm still going through the process I have a few things I can mention.
Firstly, have a plan, and then realise that the plan is more like a framework. I've changed around bits of my plan already, but the things I've intended to cover, I've mostly covered. The order and priorities have changed, but overall, I can say whether or not I'm on track. The very nature of research will present you with new things you never expected and that will force changes, but be aware of what you are spending your time on. Make sure you keep plenty of time spare for writing. In my case, I've got around 4 months penciled down, which might not even be enough.
Secondly, regular contact with folks, especially your supervisor, is important. I work remotely - very remotely! I live in Washington DC but I'm still talking to my supervisor in London every couple of weeks with some news. It helps keep you on the straight and narrow and reminds you that you are not alone. In fact, this is very important to remember. I've had help from my wife, my friends and even people I've never met at the London Biohackspace, so keep in touch with folk.
Having the right tools is important, so long as you are spending time using the tools to get the work done! I've setup an AI machine that I've since not messed with; I can rely on it to just work. I've not upgraded tensorflow or any of the libraries on it and I won't do until the job is done. I use Zotero to keep all my references in check, Latex for all my writing and I use a timer to record how long I'm spending each week on research. I'm a big fan of Trello for keeping track of the things you need to be doing and any ideas that come into your head.
Finally, choose your project wisely. I went around at least 6 different supervisors, asking them about the projects that interested me. Not only that, I spoke with a couple of friends (who are both doctors in computer science) about which project sounded right for me, and I'm really glad I did! I've ended up with a project I truly enjoy and perhaps, that's the most important thing. That way, you'll get it finished and to a high standard. Love of the project is needed to get through the tough stages (and I've already had a couple of these). Ask people you trust what they think of your options. You'll likely make a better choice.
In the next post, I'll talk a bit about the different kinds of neural networks we can make: from conv nets to LSTMs. I'll go into a little more detail about the various tests and algorithms we use to assess biological structures and what problems I've encountered on the way. I'll also talk a little about Jupyter notebooks and how we can make science a bit more accessible.
